
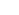


The aim of the Laboratory is to study all molecular biological systems using physical, chemical, and computational principles in order to understand such systems and to develop mathematical and computational models. This will make possible the establishment of predictive biosystems models, which can be used in applications such as the development of new drugs and innovative diagnosis. The main research areas include the prediction/analysis of the structure and function of proteins, computational study of protein interactions, computational protein design, and computational drug discovery.
Protein Bioinformatics
In order to develop the accurate prediction systems for proteins, it is essential to develop several key prediction methods. Recently, we developed a remote homolog detection algorithm with the highest sensitivity, a prediction method for hot spots in protein interaction interfaces, new methods for detecting correlated mutations in proteins and optimizing sequence alignment using correlated mutation information, phosphorylation sites prediction method, discovering disease-causing mutations, and protein domain-peptide binding energy calculation method which demonstrated the best performance in the RECOMB DREAM4 challenge: Prediction of Peptide Recognition in SH3 Domains. By combining all these developments, we aim to create a total prediction system for proteins which can be utilized for diverse applications such as protein design and drug discovery.
Computational Protein Design
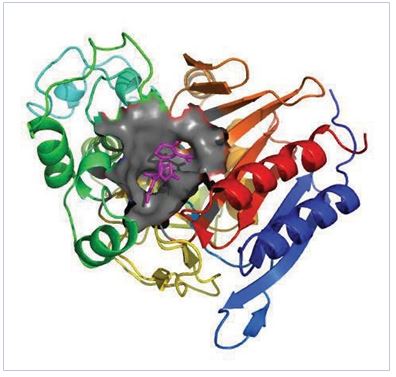
In protein design, we are trying to create new non-natur al pr ot eins with mor e desir abl e properties, for example, more efficient enzymes, stronger antibodies, and more specific transcription factors, etc. Experimental ways t o design a new 2016 Annual Report
protein is a very inefficient process relying on serendipity, requiring enormous amount of time and effort. Teamed up with experimental groups, we are developing a new computational method and strategy for protein design by combining sequence analysis, protein structure prediction, protein-ligand interaction, protein-protein interaction, protein structure analysis, and molecular dynamics simulation.
Computational Drug Discovery
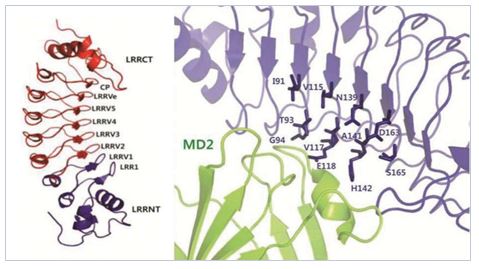
Integrating the biological networks and a variety of information can provide entirely new opportunity to develop new drugs. We are also studying physico-chemical and binding properties, such as ADME/Tox properties and protein-ligand interaction property. We are participating as a major player in the international research consortium for S. pombe mutant library project, whose main goal is to develop experimental and computational tools for systems biology and drug discovery. PBIL is primarily responsible for most of the bioinformatics works required for the project.
Computational Epigenomics
Finally, teamed with experimental groups, we aim to develop computational tools for studying epigenomics using next-generation sequencing data. In one such successful collaboration, we were able to discovery several interesting new findings about H2B monoubiquitylation (H2Bub1), one of histone modifications that are important for regulating gene regulation.
- 1. Hong S, Kim D, "Computational characterization of chromatin domain boundary-associated genomic elements.", Nucleic Acids Res. 2017 Oct 13;45(18):10403-10414.
- 2. Hong, S, Kim, D, “Library of binding protein scaffolds (LibBP): a computational platform for selection of binding protein scaffolds”, BIOINFORMATICS, vol.32, no.11, pp.1709~1715, (2016)
- 3. S-C. Lee, K. Park, J. Han, J. Lee, H.J. Kim, S. Hong, W. Heu, Y. J. Kim, J-S. Ha, S-G Lee, H-K Cheong, Y.H. Jeon, D. Kim, and H-S. Kim, “Design of a binding scaffold based on variable lymphocyte receptors of jawless vertebrates by module engineering”, PNAS, 109(9):3299-3304 (2012).
- 4. I. Jung, S-K Kim, M. Kim, Y-M. Han, Y.S. Kim, D. Kim,D. Lee, “H2B monoubiquitylation is a 5 -enriched active transcription mark and correlates with exon-intron structure in human cells”, Genome Research, 22:1026-1035 (2012).
- 5. D.-U. Kim, J. Hayles, D. Kim, et al., “Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe”, Nature Biotechnology. 28:617-623 (2010)
- 6. K. Cho, D. Kim, D. Lee, “A feature-based approach to modeling protein-protein interaction hot spots”, Nucleic Acids Research, 37:2672-2687 (2009).
- 7. K. Park, D. Kim, “Localized network centrality and essentiality in the yeast protein interaction network”, Proteomics, 9:1543-1544 (2009). (Cover article)
- 8. S. Han, D. Kim, “Inference of Protein Complex Activities from Chemical-genetic Profile and its Applications: Predicting Drug-Target Pathways”, PLoS Comp. Biol., 4(8):e1000162 (2008).
- 9. K. Park, D. Kim, “Binding similarity network of ligand”, Proteins, 71:960-971 (2008). (Faculty of 1000, Must-Read)